Peptide sequencing verification is defined as the analytical process of confirming the exact amino acid sequence and molecular identity of a peptide using mass spectrometry and complementary techniques. The process begins with intact mass measurement, then advances to tandem MS/MS fragmentation to generate b- and y-ion series that reveal the sequence order residue by residue. High-resolution instruments like Q-TOF and Orbitrap achieve mass accuracy below 5 ppm, setting the current gold standard for verification of peptide sequences. Computational tools then interpret the resulting spectral data to confirm structural correctness. For researchers working with research-grade compounds, understanding this process is the difference between confident data and costly misassignment.
How does peptide sequencing verification work?
Verification of peptide sequences follows a two-stage mass spectrometry workflow. The first stage is intact mass measurement, called MS1, which confirms the molecular weight of the intact peptide. The second stage is tandem mass spectrometry, MS/MS, which fragments the peptide and produces ions that reveal the amino acid sequence directly.
MS/MS is the definitive verification method over intact mass alone. An intact mass match confirms the molecular formula but cannot distinguish isomers or detect sequence scrambling. MS/MS breaks that ambiguity by generating a ladder of fragment ions, each differing by the mass of one amino acid residue.

Two ionization methods drive most peptide analysis techniques in 2026. Electrospray ionization (ESI) produces multiply charged ions suited to liquid chromatography coupling and complex mixtures. Matrix-assisted laser desorption/ionization (MALDI) generates singly charged ions and works well for rapid screening of purified peptides. Each ionization method feeds into the fragmentation stage, where the real sequencing work happens.
What mass spectrometry methods are used in peptide sequencing verification?
The three instrument platforms researchers rely on are MALDI-TOF, Q-TOF, and Orbitrap. Each serves a distinct role depending on the required mass accuracy and the complexity of the sample.
MALDI-TOF suits rapid identity screening. Its tolerance of ±1 Dalton is acceptable for confirming the approximate molecular weight of a purified peptide but insufficient for detecting small modifications like deamidation. Q-TOF and Orbitrap instruments achieve mass accuracy below 0.1 Da or below 5 ppm, which is the standard required to distinguish modifications that differ by fractions of a dalton.
Fragmentation method selection determines the quality of the MS/MS data. The three main approaches are:
- CID (Collision-Induced Dissociation): The most widely used method. Generates clean b- and y-ion series for standard peptides. Works well for unmodified or stably modified sequences.
- HCD (Higher-Energy Collisional Dissociation): A higher-energy variant of CID used on Orbitrap platforms. Produces richer fragment ion coverage and is preferred for phosphopeptides and other modified sequences.
- ETD/ECD (Electron Transfer/Electron Capture Dissociation): Preserves labile modifications that CID destroys. The preferred method for retaining post-translational modifications during fragmentation, particularly phosphorylation and glycosylation.
Choosing the wrong fragmentation method produces incomplete ion series. Incomplete series leave gaps in the sequence ladder, which forces researchers to rely on inference rather than direct confirmation.
Pro Tip: When working with peptides carrying labile modifications, run both HCD and ETD on the same precursor ion. Combining the two datasets gives you complementary coverage and reduces the chance of a missed modification.
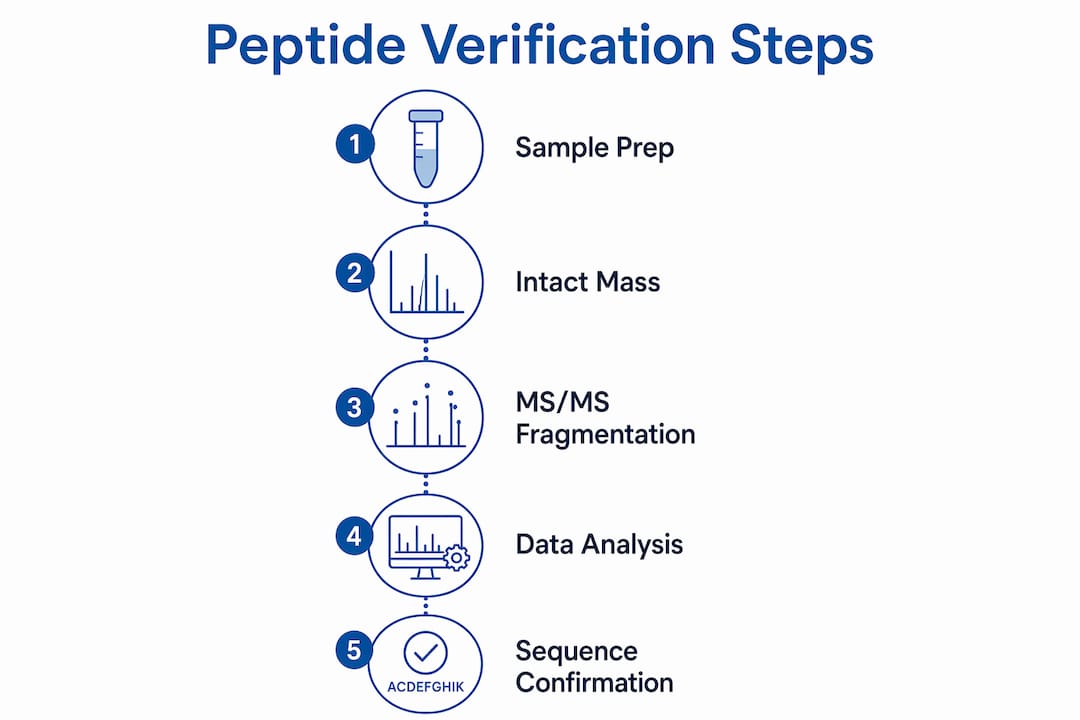
How is peptide sequence data interpreted and verified?
Spectral interpretation converts raw mass data into a confirmed amino acid sequence. The process follows a logical order, and skipping any step introduces error.
- Calculate the theoretical mass. Before running the instrument, calculate the expected monoisotopic mass of the target peptide from its sequence. This gives you the reference value for MS1 comparison.
- Match the intact mass. Compare the measured MS1 mass to the theoretical value. A match within instrument tolerance confirms the molecular formula. A mismatch signals a synthesis error, modification, or contamination.
- Assign b- and y-ions. In the MS/MS spectrum, b-ions carry the N-terminus and y-ions carry the C-terminus. The mass difference between consecutive ions in each series equals the residue mass of one amino acid. Reading these differences in order reconstructs the sequence.
- Choose between database searching and de novo sequencing. Database searching matches fragment patterns against known sequences and works fast for standard peptides. De novo sequencing reads the spectrum without a reference and is required for novel or heavily modified peptides.
- Annotate all modifications. Failure to specify modifications during MS/MS data analysis causes incorrect sequence assignments. Every known or suspected modification must be entered into the search parameters before analysis begins.
- Deconvolute ESI spectra. ESI produces multiply charged ions. Spectrum deconvolution converts these multiply charged signals into a single neutral mass, which is then compared to the theoretical value.
The computational side of peptide sequencing methods is advancing rapidly. The PowerNovo2 deep learning framework speeds up de novo sequencing by 4.3 times compared to traditional autoregressive models without sacrificing accuracy. That speed matters when researchers are processing large batches of novel peptides with no reference database entry.
Pro Tip: Always run a blank injection between samples when using LC-MS/MS. Carryover from a high-concentration sample can appear as a false positive in the next run, leading to a misidentified sequence.
What are the industry standards and tolerance levels for peptide sequencing verification in 2026?
Tolerance levels define whether a mass measurement counts as a confirmed match or a flag for further investigation. The table below summarizes the current benchmarks.
| Instrument type | Mass accuracy | Typical use case |
|---|---|---|
| MALDI-TOF | ±1 Da | Rapid purity screening of purified peptides |
| Q-TOF | <0.1 Da / <5 ppm | High-confidence sequence confirmation |
| Orbitrap | <0.1 Da / <5 ppm | Modified peptides, complex mixtures |
A discrepancy larger than the instrument tolerance signals a problem. A +16 Da shift indicates oxidation; a +1 Da shift indicates deamidation. Both are common degradation events that high-resolution MS can detect and characterize individually. Dismissing these shifts as instrument noise is a critical error. They represent real chemical changes that alter the peptide’s behavior in a research context.
Purity standards complement mass accuracy requirements. The accepted purity benchmark by HPLC-UV is greater than 95% area, with MS used to characterize each detected impurity individually. A peptide that passes the mass check but fails the purity threshold still requires further purification before use in quantitative research.
Full sequence coverage in MS/MS data is the final quality benchmark. Partial coverage, meaning gaps in the b- or y-ion ladder, leaves portions of the sequence unconfirmed. Researchers should require complete or near-complete ion series coverage before accepting a verification result as definitive.
What practical steps do researchers follow for accurate verification?
A reliable verification workflow integrates chromatography, mass spectrometry, and computational analysis in a defined sequence. Skipping steps or reordering them introduces compounding errors.
The workflow starts before the instrument is turned on. Calculate the theoretical monoisotopic mass and identify all expected modifications. This preparation step prevents misassignment at the interpretation stage.
LC-MS/MS provides both specificity and sensitivity for accurate sequence verification. Coupling high-performance liquid chromatography (HPLC) to the mass spectrometer separates peptide components before ionization. Separation reduces ion suppression, which is the interference that occurs when multiple peptides ionize simultaneously and compete for detector signal.
Key steps in a complete verification workflow:
- Step 1: Theoretical mass calculation. Use the peptide sequence and known modifications to calculate the expected monoisotopic mass.
- Step 2: HPLC purity assessment. Run HPLC-UV to confirm purity above 95% before MS analysis. Impure samples complicate spectral interpretation.
- Step 3: MS1 intact mass measurement. Confirm the molecular weight matches the theoretical value within instrument tolerance.
- Step 4: MS/MS fragmentation. Select the fragmentation method based on peptide properties. Use CID or HCD for standard peptides; use ETD for labile modifications.
- Step 5: Spectral interpretation. Assign b- and y-ions, annotate modifications, and confirm full sequence coverage.
- Step 6: Cross-check with a certificate of analysis. Compare your results against the COA documentation provided with the compound. Discrepancies between your data and the COA require investigation before proceeding.
Researchers working with verified research peptides should confirm that the supplied COA includes both intact mass data and MS/MS sequence confirmation. A COA that reports only HPLC purity without MS data is incomplete for sequencing verification purposes.
Key takeaways
Peptide sequencing verification requires intact mass measurement, MS/MS fragmentation, and computational interpretation working together to confirm amino acid sequence identity with confidence.
| Point | Details |
|---|---|
| Two-stage MS workflow | MS1 confirms molecular weight; MS/MS confirms the amino acid sequence via fragment ions. |
| Fragmentation method matters | CID suits standard peptides; ETD preserves labile modifications that CID destroys. |
| Mass accuracy thresholds | MALDI-TOF tolerates ±1 Da; Q-TOF and Orbitrap require <5 ppm for reliable verification. |
| Modification annotation is mandatory | Unspecified modifications cause false sequence assignments during MS/MS data analysis. |
| Purity and sequence coverage together | HPLC-UV purity above 95% and complete MS/MS ion coverage are both required for a valid result. |
What I’ve learned from years of watching sequencing verification go wrong
The most common mistake I see researchers make is treating intact mass confirmation as the finish line. A molecular weight match feels satisfying. The number lines up, the box gets checked, and the experiment moves forward. But intact mass alone cannot tell you whether two amino acids are transposed, whether a modification is present at the wrong residue, or whether a synthesis byproduct shares the same nominal mass as your target peptide.
The fragmentation step is where verification actually happens. And the fragmentation step is where most shortcuts get taken. Researchers use CID by default because it is the instrument default, not because it is the right choice for their peptide. For a phosphopeptide or a glycopeptide, CID strips the modification before the fragment ions are recorded. The resulting spectrum looks clean but is missing the modification data entirely. ETD or ECD would have preserved it.
The rise of deep learning tools like PowerNovo2 is genuinely changing the speed of de novo sequencing. Faster computation means researchers can verify novel peptides in batches that would have taken days just a few years ago. But speed amplifies errors when the input parameters are wrong. A fast wrong answer is worse than a slow right one. The computational tool is only as good as the modification annotations and search parameters the researcher provides.
My practical advice: treat every mass discrepancy as meaningful until proven otherwise. A +1 Da shift is not instrument noise. It is a deamidation event that may affect your peptide’s charge state, binding affinity, and stability. Confirm it, characterize it, and decide whether the compound is still fit for your research purpose. That discipline separates reliable data from data that looks reliable.
— Paul
Novatherix Laboratories: verified peptides for confident research
Researchers who need compounds they can trust from the first injection rely on Novatherix. Every peptide in the Novatherix catalog undergoes rigorous third-party analytical testing, with results documented in a full certificate of analysis that includes intact mass data, MS/MS sequence confirmation, and HPLC-UV purity above 99%.
Novatherix ships verified research peptides across the U.S. with fast turnaround and complete documentation. Whether you are working with metabolic peptides, cellular signaling compounds, or complex blends, the Novatherix product catalog gives you the analytical transparency your research demands. Every COA is third-party verified, not self-reported.
FAQ
What is peptide sequencing verification?
Peptide sequencing verification is the process of confirming a peptide’s exact amino acid sequence using mass spectrometry. It combines intact mass measurement (MS1) with tandem fragmentation (MS/MS) to produce direct sequence evidence.
What is the mass accuracy standard for peptide verification in 2026?
MALDI-TOF instruments operate within ±1 Da tolerance; high-resolution platforms like Q-TOF and Orbitrap achieve below 5 ppm. High-resolution instruments are required when detecting small modifications like oxidation (+16 Da) or deamidation (+1 Da).
What do b-ions and y-ions tell you in MS/MS analysis?
B-ions contain the N-terminal portion of the peptide and y-ions contain the C-terminal portion. The mass difference between consecutive ions in each series equals the residue mass of one amino acid, allowing direct sequence reading.
Why is fragmentation method selection important?
Different fragmentation methods generate different ion types and preserve different modifications. CID and HCD work well for standard peptides, while ETD and ECD are required to retain labile modifications like phosphorylation without destroying them during fragmentation.
What purity level is required before MS sequencing verification?
The accepted standard is greater than 95% purity by HPLC-UV area before MS analysis. Impure samples produce complex spectra that complicate ion assignment and increase the risk of false sequence identification.
Recommended
- KissPeptin-10 – Novatherix Laboratories
- Novatherix Laboratories | Research Peptides, COAs, Third Party Testing & Fast Shipping
- Research – Novatherix Laboratories