Purity verification is the process of confirming that research materials are authentic, uncontaminated, and meet the quality standards required for reliable clinical research outcomes. The role of purity verification in clinical research extends far beyond a regulatory checkbox. It determines whether your study inputs are what they claim to be, whether your data can be reproduced, and whether your findings will hold up to scrutiny. Global frameworks including ICH Q3A/B, Q3C, Q3D, and M7 set the floor for impurity control, and Novatherix Laboratories operates well above that floor with third-party verified, 99%+ purity compounds. What follows is a direct, technical breakdown of why purity verification defines research quality.
What regulatory frameworks govern purity verification in clinical research?
Purity verification in clinical research is shaped by a set of binding international guidelines, not voluntary best practices. The ICH suite of guidelines, specifically Q3A/B for drug substance and drug product impurities, Q3C for residual solvents, Q3D for elemental impurities, and M7 for mutagenic impurities, defines the categories of contaminants that must be profiled and controlled.
ICH M7, combined with USP <1469>, specifically addresses nitrosamine risk assessment. Nitrosamines are potent genotoxic impurities that can form during synthesis or storage. Impurity profiling under M7 covers organic, inorganic, residual solvent, and genotoxic impurity classes, each with distinct acceptable limits tied to clinical exposure levels.

ICH Q7 adds another layer by requiring 100% identity testing of all incoming active pharmaceutical ingredients. This mandate exists because mix-ups, adulteration, and mislabeling are real failure modes, not theoretical ones. Identity testing methods under Q7 include Fourier-transform infrared spectroscopy (FTIR), near-infrared spectroscopy (NIR), and Raman spectroscopy, all of which provide rapid, independent confirmation of material identity.
These regulations directly shape clinical research protocols in three ways:
- Supplier qualification: Researchers must document supplier history, audit results, and material specifications before a compound enters a study.
- Impurity limits by phase: Acceptable impurity thresholds tighten as a study progresses from early-phase to late-stage trials, requiring updated testing at each transition.
- Documentation requirements: Every batch used in a study must carry traceable, batch-specific records that satisfy both the originating lab and any regulatory authority reviewing the data.
Ignoring these frameworks does not just create a compliance gap. It creates a scientific one. Data generated from materials that were never properly characterized cannot be reliably interpreted or reproduced.
How does purity testing enhance the integrity of research outcomes?
Purity assessment in trials directly controls one of the most overlooked sources of variability: the starting material. Variability in ingredient purity impacts outcome reliability and regulatory confidence in clinical studies. A compound dosed at a nominal concentration but carrying 3–5% unknown impurities is not the compound the protocol describes.
Standardizing study inputs through verified purity achieves four concrete benefits:
- Dose accuracy: When a compound is confirmed at 99%+ purity, researchers can calculate actual administered doses with confidence. Impure materials introduce silent dosing errors.
- Reduced confounding: Unidentified impurities can produce biological effects that mimic, mask, or amplify the response under study. Verified purity removes that confounder.
- Reproducibility: A study repeated with the same compound at the same verified purity will produce comparable results. Studies using uncharacterized materials cannot be reliably replicated.
- Regulatory acceptance: Regulatory reviewers assess the quality of materials used in a study. Documented purity verification strengthens the credibility of submitted data.
Certificates of Analysis (COAs) are the primary documentation tool for purity in research settings. However, a COA is only as reliable as the lab that produced it. Batch-specific COAs from ISO 17025 accredited labs are the standard that protects research data. Generic templates or COAs from unaccredited sources can invalidate an entire dataset.
Pro Tip: Always request a batch-specific COA, not a generic product specification sheet. The batch number on the COA must match the batch number on the material you received. If they do not match, the document is not valid for your study.
The financial argument reinforces the scientific one. Testing incoming raw materials costs significantly less than managing failed batches or conducting market withdrawals. Catching a purity failure before a study begins is always cheaper than discovering it after data collection.
What methods are used to verify purity in research materials?
Purity verification processes fall into two functional categories: identity confirmation and impurity quantification. Each serves a distinct purpose, and selecting the right method depends on material criticality, study phase, and regulatory context.
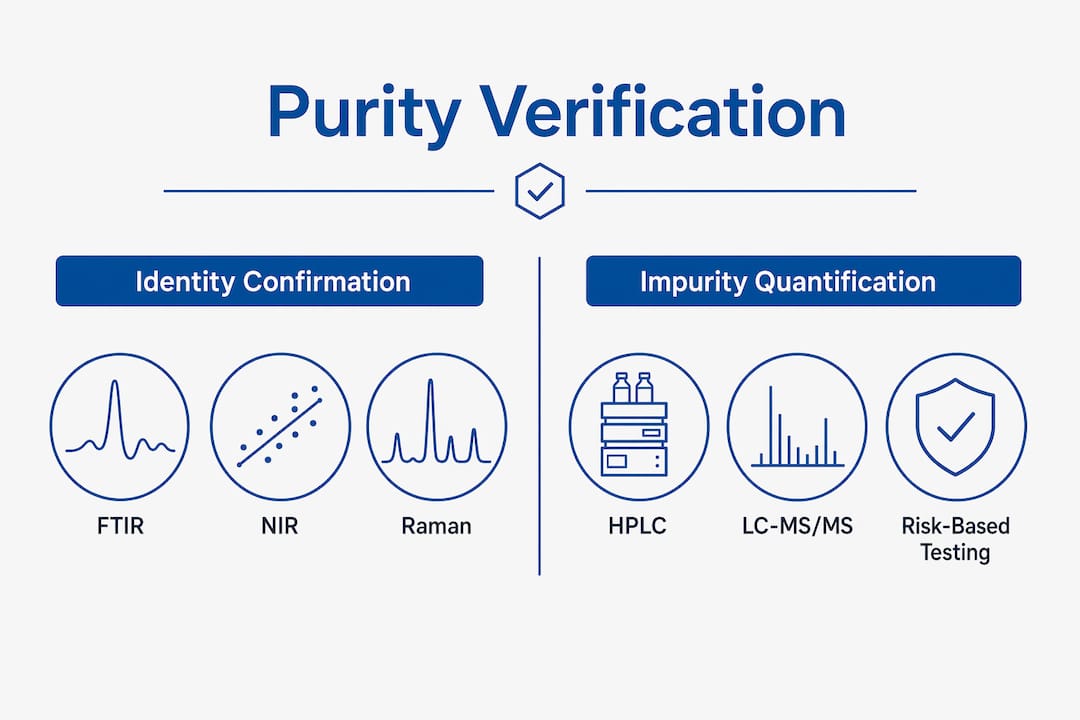
Identity confirmation techniques
FTIR, NIR, and Raman spectroscopy are the standard tools for identity testing. Each generates a spectral fingerprint that confirms a material matches its reference standard. FTIR is the most widely used because it works across a broad range of compound classes. NIR offers speed advantages for high-throughput screening. Raman spectroscopy excels with aqueous solutions and materials in sealed containers, since it does not require sample preparation.
Impurity quantification methods
| Method | Primary use | Key advantage |
|---|---|---|
| HPLC | Organic impurity profiling | High resolution, widely validated |
| GC | Residual solvent analysis | Sensitive to volatile compounds |
| LC-MS/MS | Genotoxic and trace impurities | Extremely low detection limits |
| NMR | Structural confirmation | Identifies unknown impurities |
LC-MS/MS is the method of choice for genotoxic impurities like nitrosamines because it achieves the parts-per-billion sensitivity that ICH M7 limits require. NMR adds structural detail that chromatographic methods alone cannot provide.
Method selection also depends on development stage. Qualified versus validated analytical methods serve different purposes. Qualified methods are acceptable for early-phase work. Validated methods, developed under ICH Q2(R2) and Q14 frameworks, are required for later-stage clinical trials. Using a qualified method where a validated one is required creates a gap that regulators will flag during review.
A risk-based testing strategy determines which materials receive full testing batteries and which receive reduced panels. Material criticality, intended use, and supplier track record all feed into that decision. Not every incoming material needs the same depth of analysis, but critical materials with direct impact on study outcomes require the most thorough characterization.
Pro Tip: Verify the accreditation status of any laboratory performing your purity testing. ISO 17025 accreditation is the minimum standard for a lab whose results will appear in regulatory submissions or peer-reviewed publications.
What challenges exist in implementing purity verification for clinical research?
Clinical research quality control around purity is more complex than running a single test and filing the result. Several persistent challenges reduce the reliability of purity data when researchers do not account for them.
- COA over-reliance: Many researchers accept a supplier’s COA without independent verification. A COA confirms what the supplier measured, not what you received. Degradation during shipping, storage failures, or simple documentation errors can all create a gap between the COA and the actual material.
- Lab variability: Purity results for the same compound can differ between labs due to differences in reference standards, instrument calibration, and method parameters. Comparing results across studies requires knowing which method each lab used.
- Genotoxic impurity complexity: Nitrosamines and other mutagenic impurities require specialized methods and risk assessments that many standard testing panels do not include. Researchers working with compounds that have nitrosamine formation potential need to specifically request M7-compliant testing.
- Phase-appropriate limits: Impurity thresholds that are acceptable in a Phase I study may not be acceptable in Phase III. Researchers who set limits once and do not revisit them as a study progresses create a compliance gap that compounds over time.
- Emerging monitoring tools: Advances in impurity control now include Process Analytical Technology (PAT), Real-Time Release Testing (RTRT), and AI-driven impurity prediction. These tools shift purity control from reactive detection to proactive prevention. Most research programs have not yet integrated them, which means they are still catching problems after they occur rather than before.
The gap between what a COA states and what independent testing reveals is where research integrity most often breaks down. Closing that gap requires treating purity verification as an ongoing process, not a one-time check at material receipt.
Key Takeaways
Purity verification is a continuous quality control process that directly determines the validity, reproducibility, and regulatory credibility of clinical research data.
| Point | Details |
|---|---|
| Regulatory frameworks set the floor | ICH Q3A/B, Q3C, Q3D, M7, and Q7 define mandatory impurity profiling and identity testing requirements for research materials. |
| COAs require independent verification | Batch-specific COAs from ISO 17025 accredited labs are the only reliable documentation standard for research-grade purity. |
| Method selection must match study phase | Qualified methods suit early-phase work; validated methods per ICH Q2(R2) and Q14 are required for later-stage clinical trials. |
| Risk-based testing improves efficiency | Material criticality and supplier history determine testing depth, focusing resources where purity failures carry the highest consequences. |
| Emerging tools shift control upstream | PAT, RTRT, and AI-driven prediction move impurity management from reactive detection to proactive prevention. |
Why purity verification deserves more than a line item in your protocol
Researchers tend to treat purity verification as an administrative task. You request a COA, file it, and move on. I have seen this approach create problems that only surface months later, when a study’s data cannot be reproduced and no one can explain why.
The uncomfortable reality is that a COA from an unaccredited lab, or one that is not batch-specific, is not evidence of purity. It is a document that looks like evidence. The distinction matters enormously when your data is under review.
What I have found actually works is treating purity verification the same way you treat your primary endpoint measurement: with defined methods, documented acceptance criteria, and independent confirmation. That means specifying in your protocol which analytical methods are required, which lab accreditation standards apply, and what happens if a material fails incoming testing. It means building purity verification into your study design, not bolting it on afterward.
The regulatory frameworks, ICH Q3A/B, Q3C, Q3D, M7, and Q7, exist because impurity failures have caused real harm in real studies. They are not bureaucratic overhead. They encode decades of hard lessons about what happens when material quality is assumed rather than confirmed.
Researchers who adopt a risk-based purity strategy and pair it with rigorous documentation consistently produce more reproducible data and face fewer regulatory questions. That is not a coincidence. Purity verification is not optional infrastructure. It is the foundation that everything else rests on.
— Paul
Novatherix Laboratories: verified purity for serious research
Researchers who need compounds they can trust from the first vial to the last data point will find that Novatherix Laboratories is built around that requirement. Every compound in the Novatherix catalog undergoes rigorous third-party analytical testing, with results documented in batch-specific COAs that meet the standards researchers and regulatory reviewers expect.
Novatherix delivers 99%+ purity research-grade compounds with fast U.S. shipping and full documentation transparency. Whether you are sourcing peptides for cellular research or metabolic studies, the Novatherix research collections give you verified starting materials backed by real analytical data. Every Certificate of Analysis is batch-specific and traceable, so your purity documentation holds up at every stage of your work.
FAQ
What is purity verification in clinical research?
Purity verification is the process of confirming that research materials match their stated identity and meet defined impurity limits. It uses analytical methods like HPLC, FTIR, and LC-MS/MS to characterize compounds before they enter a study.
Why are batch-specific COAs required for research materials?
Generic COAs do not confirm the quality of the specific batch you received. Batch-specific COAs from ISO 17025 accredited labs are the only documents that provide traceable, legally defensible evidence of purity for a given lot.
What is the difference between qualified and validated analytical methods?
Qualified methods demonstrate fitness for purpose and are acceptable in early-phase research. Validated methods under ICH Q2(R2) meet a higher evidentiary standard and are required for later-stage clinical trial submissions.
How do impurity limits change across clinical trial phases?
Impurity thresholds tighten as trials advance from Phase I to Phase III because cumulative patient exposure increases. Limits acceptable in early-phase studies may require tighter controls once a compound moves into longer-duration or larger-scale trials.
What makes a purity testing strategy risk-based?
A risk-based strategy assigns testing depth based on material criticality, intended use, and supplier track record. Critical materials with direct impact on study outcomes receive full analytical panels, while lower-risk materials may require reduced testing.
Recommended
- Novatherix Laboratories | Research Peptides, COAs, Third Party Testing & Fast Shipping
- Research Preparation & Reconstitution Guide – Novatherix Laboratories
- KPV – Novatherix Laboratories